
【文献阅读】DNA language models are powerful predictors of genome-wide variant effects
写在前面:很久之前的JC记录。
1 Information of this paper
1.1 Publication time, Journal, Author
-
Received July 3, 2023; Accepted September 8, 2023
-
PNAS IF: 11.1
-
Correspondence Author: Yun S. Song (University of California, Berkeley)
-
Link:https://www.pnas.org/doi/10.1073/pnas.2311219120

1.2 Abstract (What does the paper want to do?)
-
Purpose: predict the effects of genetic variants across the entire genome
-
Model: Genomic Pre-trained Network (GPN), a model designed to learn 1)genome-wide variant effects through **unsupervised pretraining on genomic DNA sequences. Our model also successfully learns 2)gene structure and 3)DNA motifs without any supervision.
-
Methods: we train GPN on unaligned reference genomesof Arabidopsis thaliana and seven related species within the Brassicales(这里的“unaligned”意思是拟南芥和其他7个相近的物种的参考基因组没有相互比对。至于为啥要用这8种植物的基因组,是因为它们之间同源关系强,序列的保守性/非保守性也会比较相似,所以用语言模型能更好地学到这几种植物的序列特征) order and evaluate its ability to predict the functional impact of genetic variants in A. thaliana by utilizing allele frequencies from the 1001 Genomes Project and a comprehensive database of GWAS.
在拟南芥和芸苔目中七个相关物种的未比对参考基因组上训练GPN(比对的工具这个课题组后续还发了一篇文章做这件事,叫GPN-MSA,这个模型就是监督的了,因为人的基因组太大了,如果不加入任何功能基因组的数据,模型很难学习到region的特征),并利用 1001 基因组计划和 GWAS 综合数据库中的等位基因频率,评估其预测拟南芥遗传变异的功能影响的能力。
-
Results: GPN outperforms predictors based on popular conservation scores such as phyloP and phastCons(这两个保守性得分相当于这个领域的金标准). Our predictions for A. thaliana can be visualized as sequence logos in the UCSC Genome Browser.
2 Introduction
2.1 Genome-wide association studies (GWAS)
GWAS是用来检测与某些特征或疾病相关的基因位点的一种研究方法。核心挑战在于,要准确地确定哪些变异是导致某个特征的真正原因,这并不容易。原因是由于遗传位点之间存在连锁不平衡(linkage disequilibrium,LD)的现象。
LD是指,在基因组的某些区域内,不同的基因位点之间存在着较强的相关性,它们倾向于被一起遗传。这种现象会导致,与疾病相关的真正病因变异,可能与其他无关紧要的变异也存在相关性。有些变异可能位于基因的编码区域,会影响基因的功能,从而导致疾病发生,我们称这种变异为”病因变异”。但是,由于基因组内存在着联系不平衡现象,病因变异附近的其他无关紧要的变异,也可能与疾病表型存在统计相关性,所以我们在没开上帝视角的前提下,没办法区分出哪个才是真正的致病性突变。
这就需要采用精细定位(fine-mapping)策略,利用计算变异效应预测器(computational variant effect predictors)寻找真正的因果变异。这种策略对于构建准确、可移植的多基因风险评分,以及阐明潜在的生物学机制至关重要。
2.2 Interpreting noncoding variant effects
解释非编码区变异效应是另一大挑战。一种广为使用的无监督方法是训练监督模型预测功能基因组数据(如染色质可及性、转录因子结合或基因表达),然后根据变异对这些预测的干扰程度来评估其效应。但该方法的成功有赖于从多种细胞类型获取高质量功能基因组学数据,对于大多数物种而言代价高昂。
另一种方式是针对特定类型的非编码变异开发专门模型(type-specific),如预测内含子变异对剪切pattern的影响,或预测调控序列变异对DNA折叠和DNA甲基化水平的影响。但这些专门模型在检测与性状相关的罕见变异、进行精细定位或计算多基因评分时可能效果不佳,因为它们无法对基因组范围的编码和非编码变异进行综合比较和排序。
3 Results
3.1 Training a Multispecies DNA Language Model
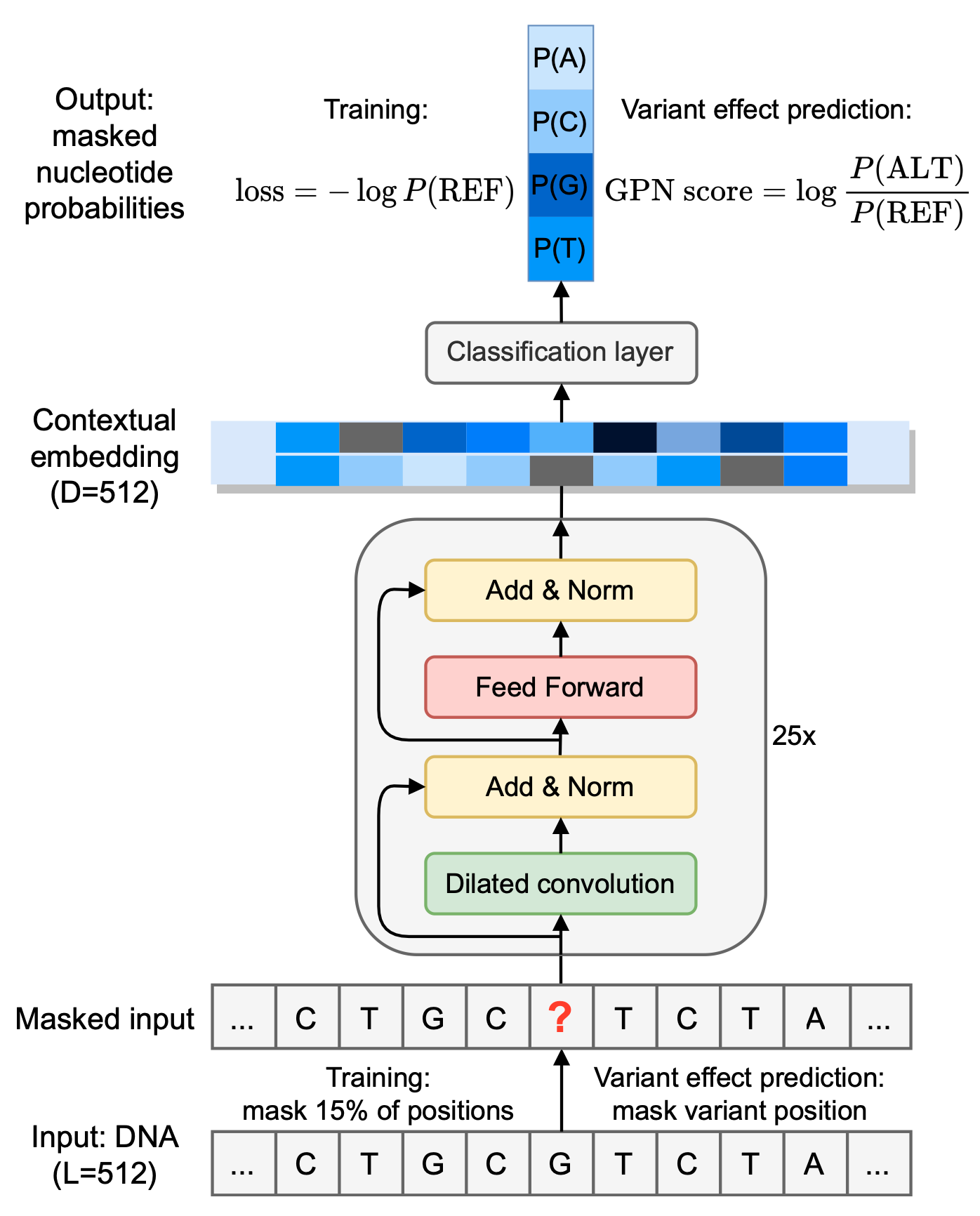
-
Training Dataset
- Unaligned reference genomes of 8 plants (A. thaliana (5 pairs of chromosomes, ~135 Mb) and 7 related Brassicales plants)
- Unsupervised model
- DNA sequences alone without the use of any functional genomic information
- Further studies: Human genome (~3.1 Gb) –> GPN-MSA (Gonzalo Benegas et al. bioRxiv, 2023)
-
Input Rules
- 512bp window & 256bp step size (fasta file)
- Not whole genome, but choose specific regions (e.g., exons, promoters) and other randomized windows equal to these regions
- Appropriately reduce the weight of prediction loss for repetitive regions
- Training: randomly masks 15% of bases
- Variant effect prediction (3.4节): mask variant position
-
About the model
-
A convolutional neural network (CNN)
- 25 convolutional blocks
-
Initial experiments:
- Convolutional models converged faster than transformer models
- Transformer虽然是自然语言模型里的宠儿,包括GPT在内的很多语言模型都在用它,但它的复杂度比CNN高,且训练所消耗的资源也很多。
-
Contextual embedding
- Use vector to represent words/bases
- 从原文提供的github中的examples/ss/basic_example.ipynb中,可以看到运行这个模型的示例,输入700base的序列,得出的embedding可视化结果如下。
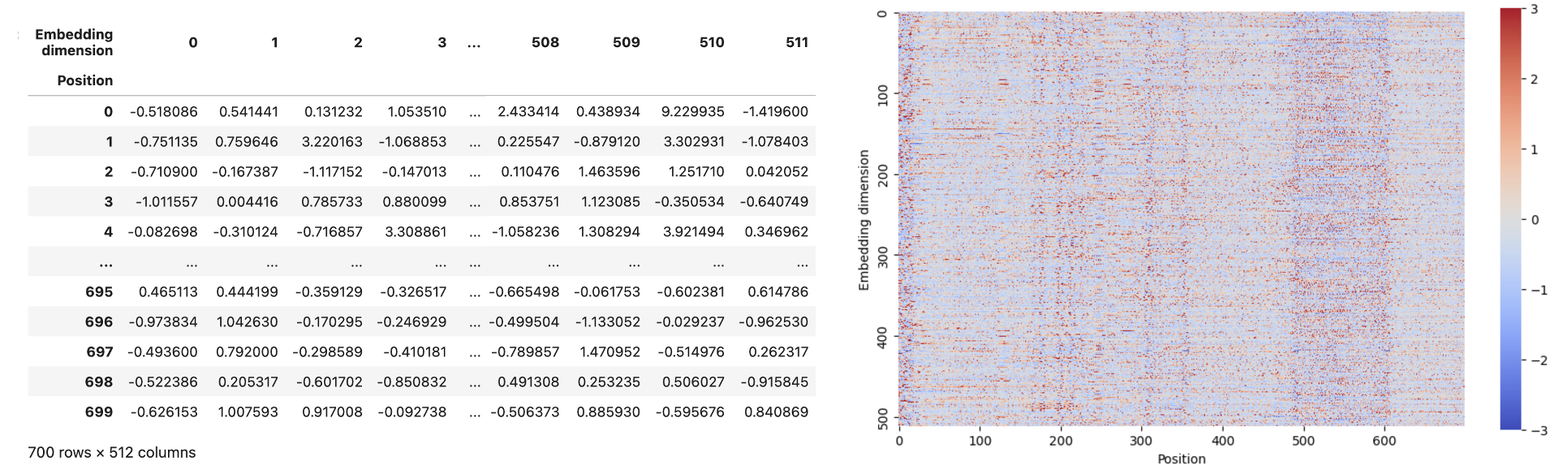
-
Training
- The model is trained on the reference sequence with the cross-entropy loss.
-
-
Output
-
Nucleotide probabilities
- P(A), P(C), P(G), P(T)
-
GPN score (3.4节)
-
$GPN score = \log \frac{P(\mathrm{ALT})}{P(\mathrm{REF})}$,$P(\mathrm{ALT})$是Alternate allele的概率,$P(\mathrm{REF})$是Reference allele的概率。
-
GPN score >0, P(ALT)>P(REF)
GPN score <0, P(ALT)<P(REF)
GPN score =0, P(ALT)=P(REF)
-
-
3.2 Unsupervised Clustering of Genomic Regions
-
Averaged GPN’s contextual embeddings of nucleotides over 100 base pair (bp) windows from the reference genome and visualized them using UMAP
- GPN, has learned to distinguish genomic regions such as intergenic, introns, coding sequences (CDS), untranslated regions (UTR), and noncoding RNA (ncRNA)
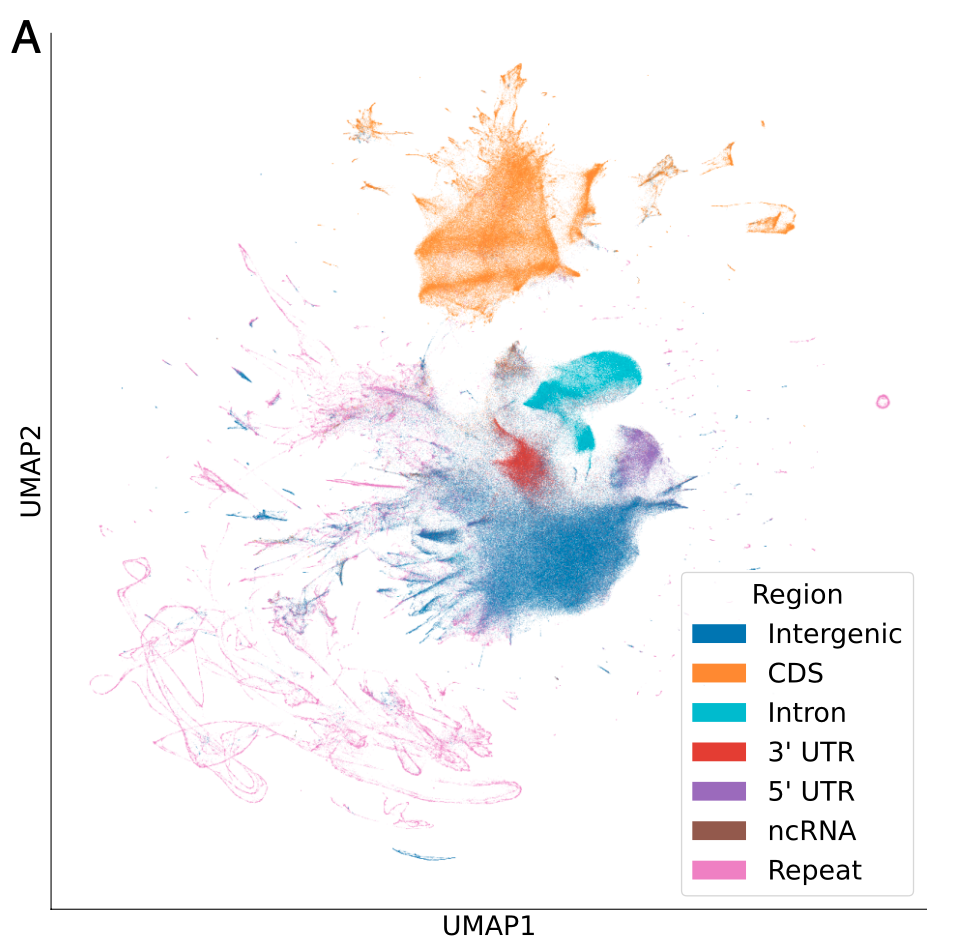
-
2 A logistic regression classifier using the averaged embeddings as features
-
The highest accuracy on CDS (96%) , the lowest on ncRNA (51%) (also the least frequent class)
-
This may be partly explained by errors in ncRNA annotation, which is especially challenging given their low expression levels and poor conservation
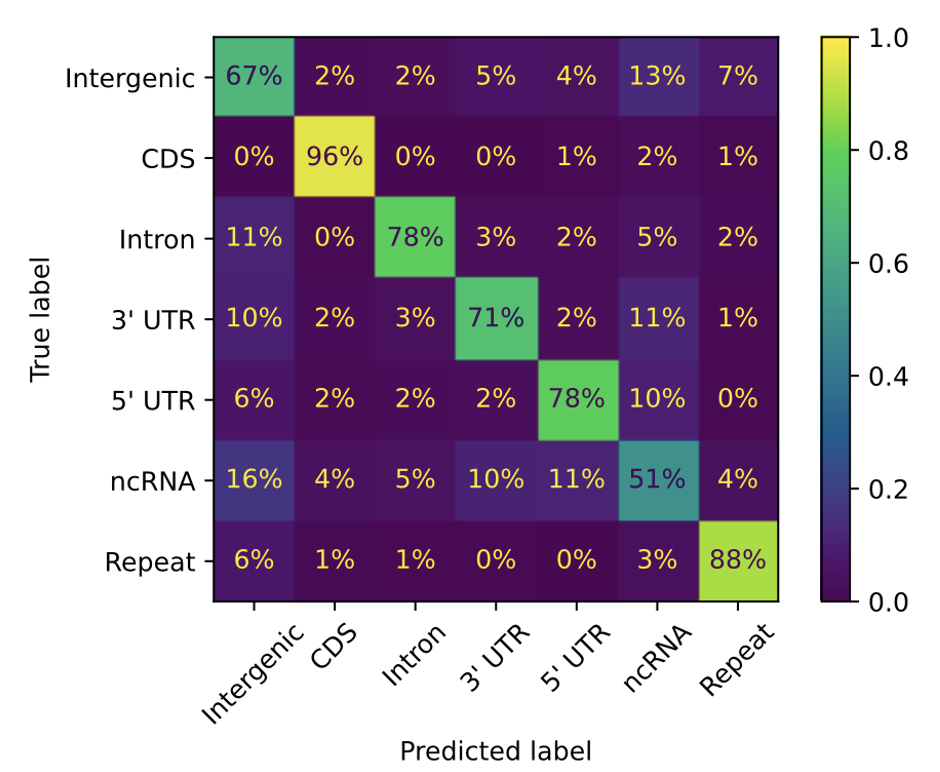
-
3.3 DNA Motifs Revealed by High-Confidence Model Predictions
-
Visualization: sequence logos (UCSC Genome Browser)
- The height of each letter is proportional to its probability, and the overall height is given by the information content, measured in bits(单个字母高度反映了单个核苷酸在该位置上的重要性,而总高度反映了该位置对于整个序列模式的重要程度。较高的总高度意味着该位置具有更高的保守性和信息含量,对于识别该序列模式更为关键。)

-
In 1Mb region (Chr5:3,500,000-4,500,000)
- The model’s prediction confidence correlates with the expected functionality of the sites
- exon>intron
- within codons: CDS2>CDS1>CDS3 (BECAUSE CDS3 usually does not affect amino acid identity ) 第三位核苷酸的改变并不一定影响所编码的氨基酸
- The model’s prediction confidence correlates with the expected functionality of the sites
-
Identify transcription factor binding sites
- Other tool: TF-MoDISco (de novo identification of transcription factor binding sites using supervised models)
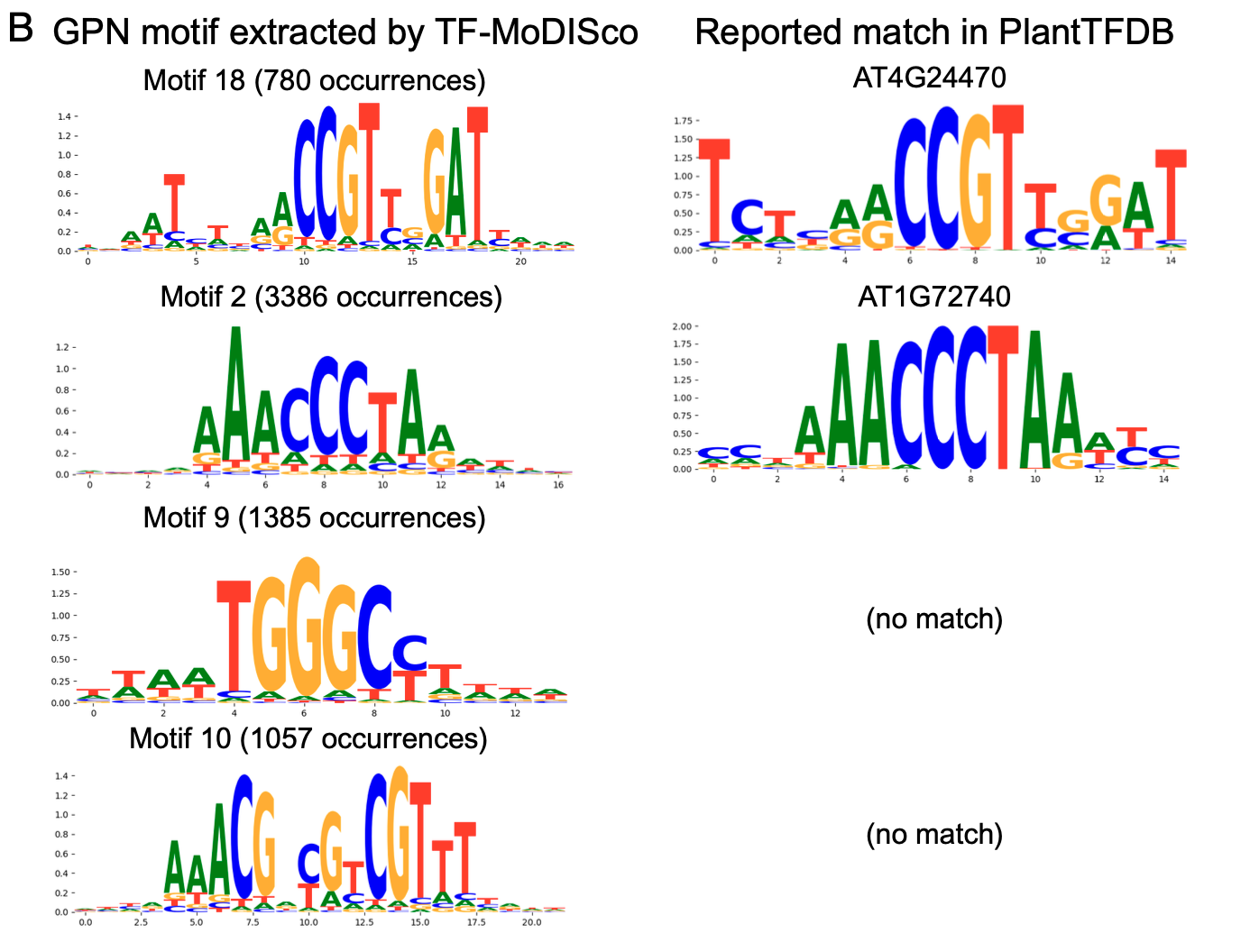
有些已确定的图案在文献中已有详细记载,但在本数据库中却没有明显的匹配,如图 中的第三个图案。有些图案可能代表以前未发现的启动子元件,如第四个图案,它是对称熵的回文图案,表明它有可能形成RNA或DNA的可变二级结构。
3.4 Unsupervised Variant Effect Prediction
-
In silico mutagenesis
Compute GPN scores for in silico mutagenesis of SNPs within a 1-Mb region and aggregated the results across variant types:
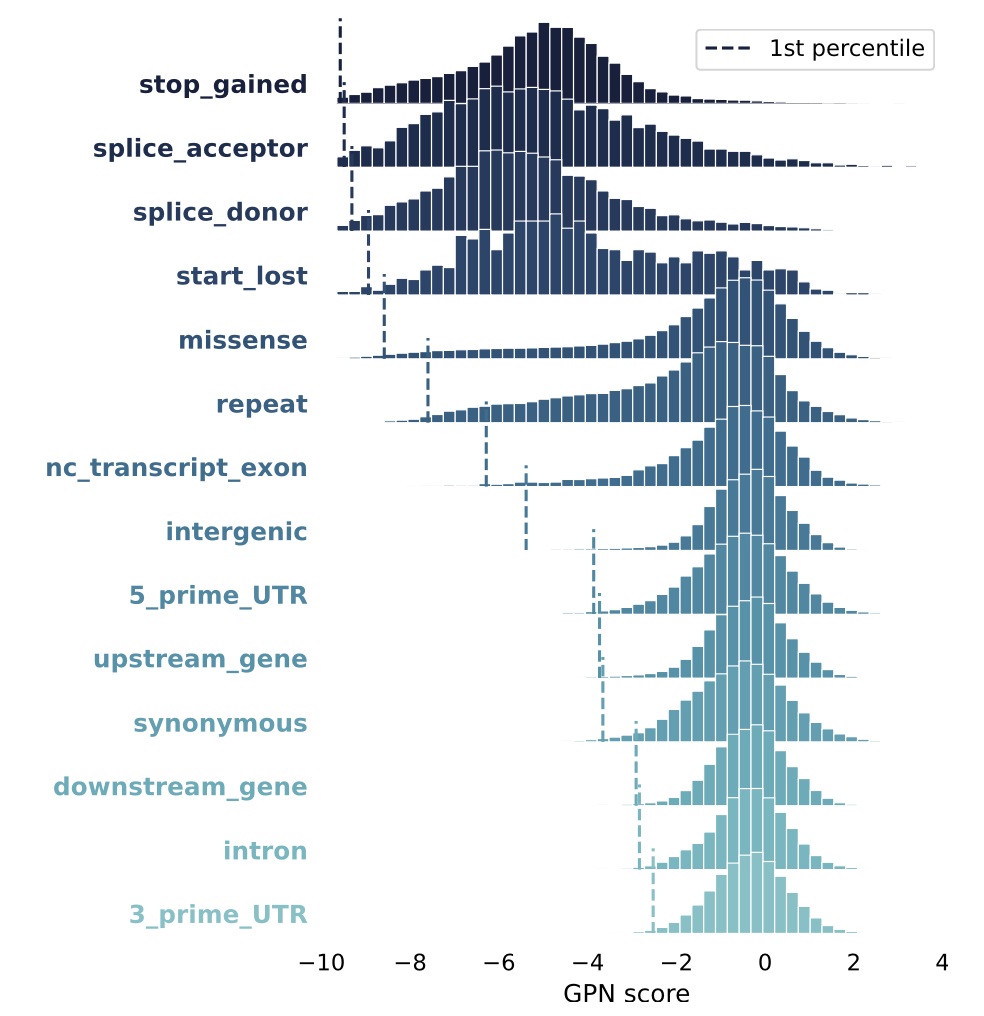
The ranking of variant types based on the lowest percentile of GPN scores is generally consistent with established notions of deleteriousness (The Ensembl Variant Effect Predictor)(Calculated consequences (ensembl.org))
-
Benchmarking using allele frequencies in 1001 Genomes
- 1001 Genomes: The 1001 Genomes Project was launched at the beginning of 2008 to discover detailed whole-genome sequence variation in at least 1001 strains (accessions) of the reference plant Arabidopsis thaliana.
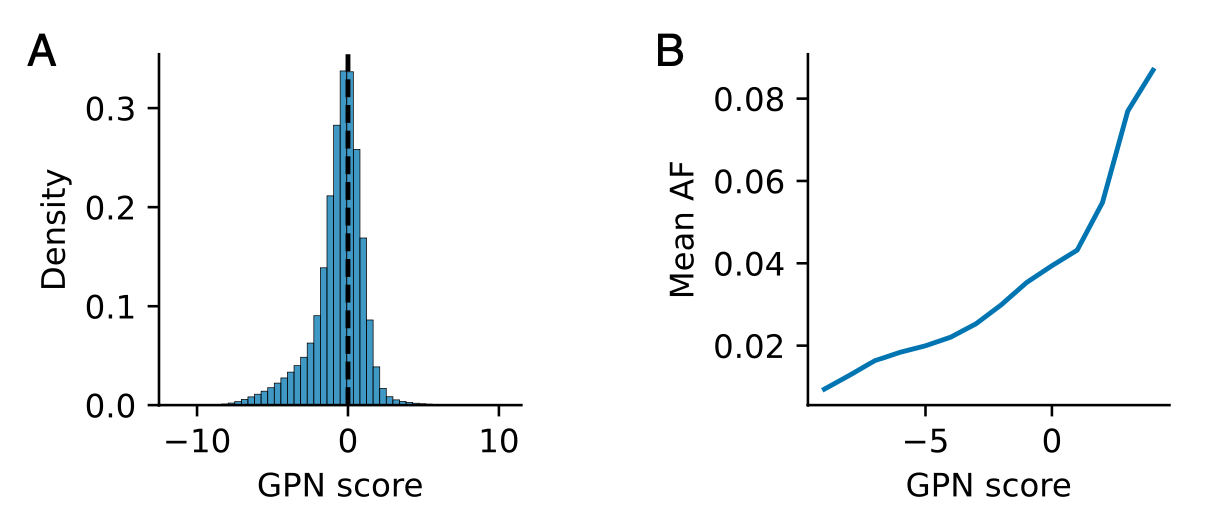
There is a heavy tail of putative functional variants with negative GPN scores (上图A部分)
虽然大多数变体的 GPN 得分是中性的,但有大量推定功能变体的 GPN 得分是负的。值得注意的是,GPN 分数较低的变体在群体中的平均出现频率较低,这表明它们可能正处于纯化选择的过程中。【GPN分数越低代表突变对生物的影响越严重】
- To evaluate the capability of identifying putative functional variants
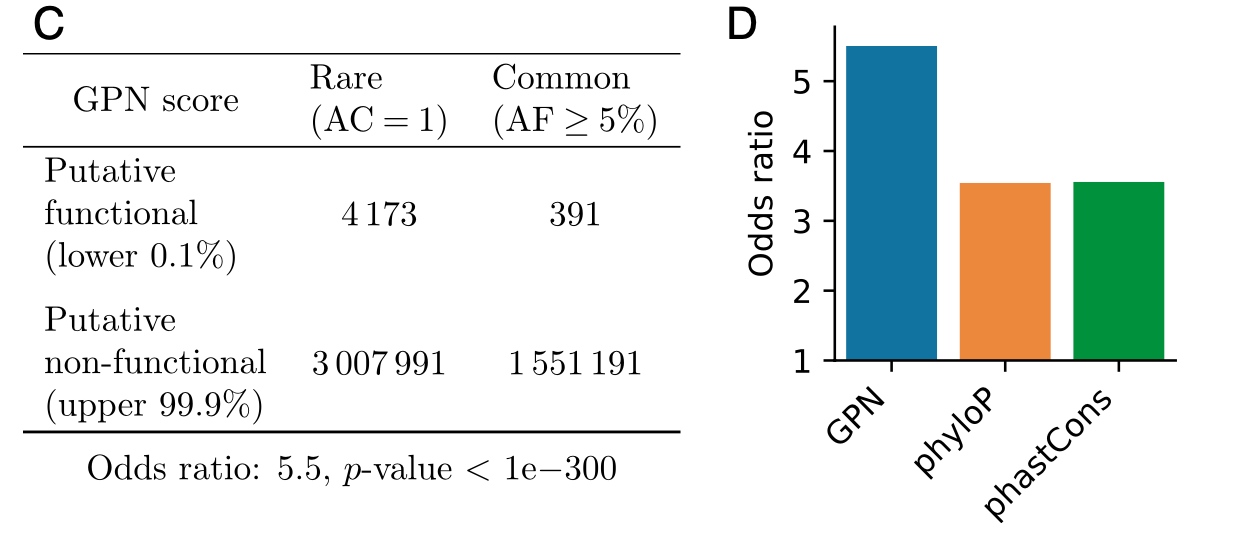
- Focusing on the tail (the lowest 0.1% of GPN scores)…被定义为 GPN 分数最低 0.1% 的推定功能 SNP 在罕见变异中富集了 5.5 倍。(AC: allele count; AF: allele frequency)
- GPN outperforms other genome-wide variant effect predictors for Arabidopsis, specifically phyloP and phastCons
- phyloP and phastCons :conservation scores derived from a broader set of 18 Brassicales species
- A notable advantage of GPN is that it is able to score variants that could not be scored by phyloP and phastCons due to unsuccessful whole-genome alignment
-
Enrichment of GWAS hits in regions with low GPN scores
(这个部分和Introduction的GWAS呼应了)
- Examined the AraGWAS Catalog (a comprehensive database of GWAS in A. thaliana)
- Hypothesis: GWAS hits may be enriched in regions with low GPN scores
- 下图是 Example window with six variants tested for association with maximum temperature in January
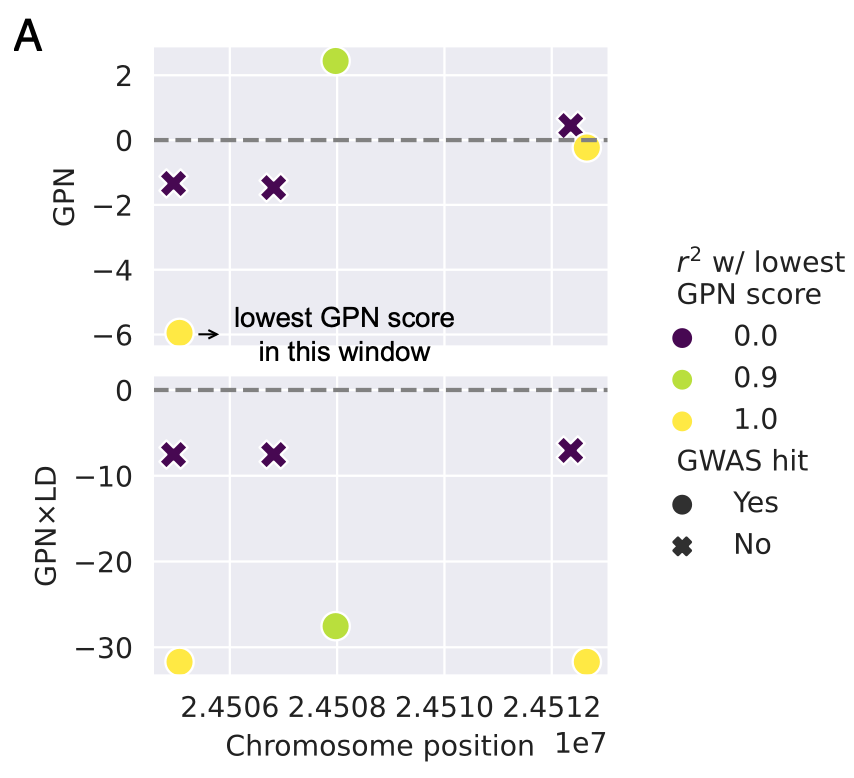
这个图曾困扰了我很久…我最后的理解是这样的。$r^2$代表两个SNP之间的关联度,$r^2$越高,代表LD越强。如果突变周围的环境不同,GPN可以给彼此处于强LD的突变(两个黄色的点)以截然不同的评分。但GWAS会给处于强LD的一个中性突变和一个功能性突变(最右边的一个黄点和一个叉号)相似的分数。
为了解决这种差异,作者又进一步设计了一个分数GPN × LD, \(\operatorname{LDScore}_i=-\sum_j r_{i j}^2 \text {. }\) \(\mathrm{GPN} \times \mathrm{LD}_i=-\sum_j\left|\mathrm{GPN}_j\right| \cdot r_{i j}^2\) 其中i和j代表不同的SNP。
该分数通过LD加权GPN分数。利用这种方法,GPN × LD有效地区分了该示例基因座中的GWAS命中与非命中(上图的下半部分)。
-
【其他的GPN × LD的优势】在整个基因组和所有性状中,GPN × LD 分数的尾部极大地富集了 GWAS 的命中率,远高于原始 GPN 分数的尾部。
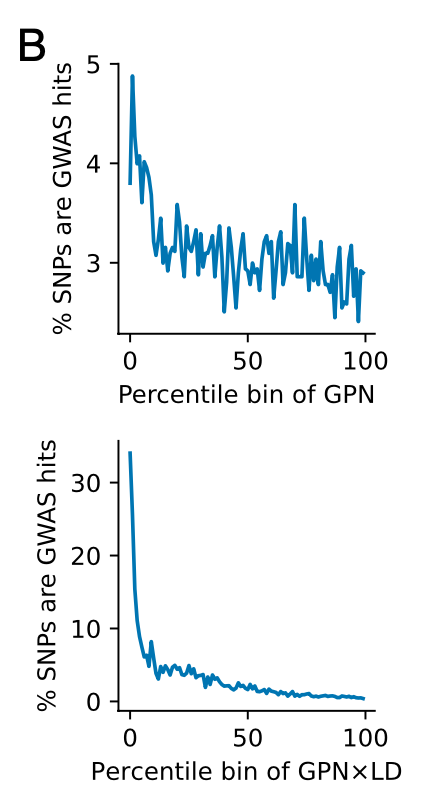
-
通过分析OR值,我们发现与 GPN × LD 分数的上部 99% 相比,GPN × LD 分数的下部 1% 的 SNP 在 GWAS 命中富集了 10.3 倍,而其他方法观察到的富集不到 7.5 倍。
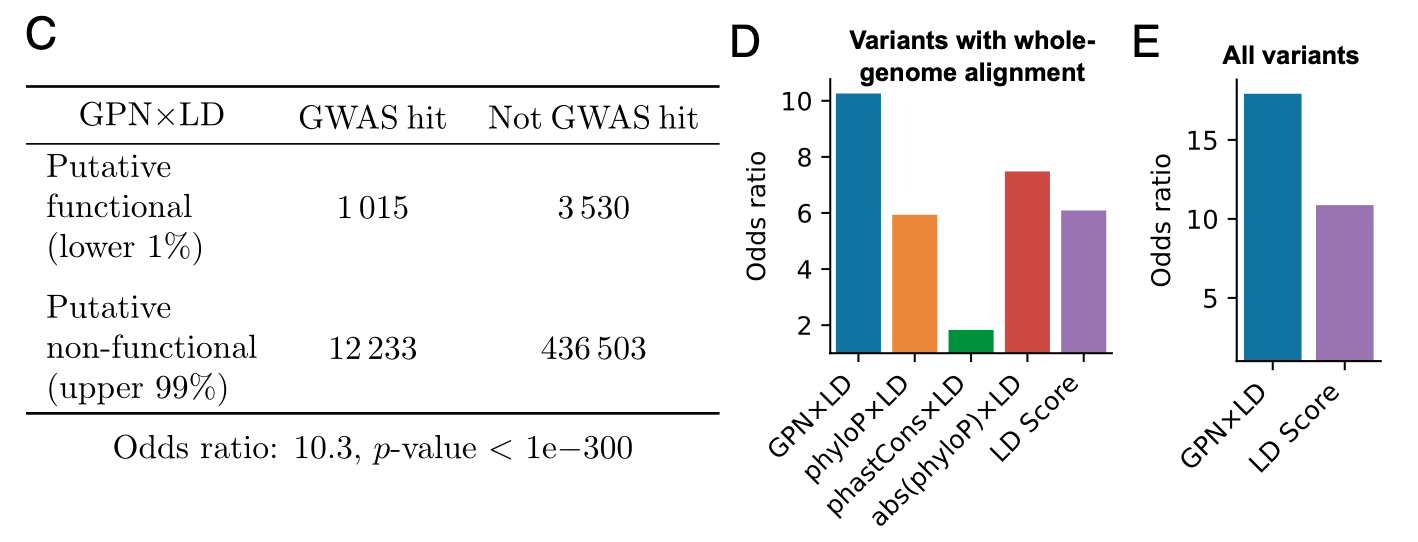
4 Discussion
- Advantages of GPN:
- GPN is genome-wide, can be trained on genomic sequence alone, and is cell-type and mechanism agnostic.
- GPN can learn from joint nucleotide distributions across all similar contexts appearing in the genome.
- GPN does not rely on whole-genome alignments, which can often have a lower quality in noncoding regions.
-
Potential research directions include examining the effects of down-weighting repeats based on their respective families or inferred age.
- To me: Common model, simple figures, but is a good story and provides a molecular biology interpretation of the results of machine learning
- 如何讲好生物故事,才是机器学习+生物最好的topic。