
【实战练习】从3'Seq数据提取polyA位点
1 背景介绍
1.1 替代性多聚腺苷化(APA)
替代性多聚腺苷酸化(Alternative Polyadenylation, APA)是真核生物基因表达调控的重要机制之一。该过程涉及到mRNA 3’末端加工的多样性,通过在不同的聚腺苷酸化(poly(A))位点进行切割和添加腺苷酸链,从而产生具有不同3’非翻译区(3’ UTR)长度和序列的mRNA变体。这种3’末端的变异可以影响mRNA的稳定性、核糖体结合以及转录后调控,进而调节蛋白质的表达水平和功能。APA可以在多个生物学过程中发挥作用,包括细胞增殖、分化、发育以及应答环境刺激等。
APA的一种特殊形式是CR-APA(Coding-Region Polyadenylation),当poly(A)信号出现在内含子区域时,会导致产生具有不同羧基端的蛋白质异构体。这不仅影响了mRNA的编码潜力,还可能引起蛋白质功能的多样化,从而增加了细胞调控复杂性。
在图1中,(A)显示了DNA中的5’端编码序列(灰色盒子)、3’非翻译区(3’ UTRs)和聚腺苷酸化位点(蓝色箭头)。(B)聚腺苷酸化是在新生mRNA上酶促合成约200个腺苷酸残基的延伸过程,在本例中使用了远端聚腺苷酸化位点。(B,C)在3’ UTR-APA中,选择近端的切割和聚腺苷酸化结果导致产生一个具有相同蛋白编码潜力但3’ UTR长度不同的mRNA。(D)当在内含子区域识别到poly(A)信号时,会通过一个称为CR-APA的过程生成具有不同羧基端的蛋白质异构体[1]。
![图 1替代性多聚腺苷酸化 (APA)[1]](https://s2.loli.net/2023/12/23/v6wirKZfR1CJQEl.png)
目前,传统 RNA-Seq、3’-Seq和sc-RNA-Seq是表征 APA 的主要方法(图2)。APA 数据库拥有从大量输入中整理出来的与 APA 和 3’ UTR 相关的信息。APA的检测需要运用生物信息学方法进行统计排序。在图2中,单细胞数据分析的生物信息学方法用红色显示[1]。
![图 2 APA的三重属性[1]](https://s2.loli.net/2023/12/23/iENROGYUBPjL7lc.png)
1.2 APA研究方法比较
在替代性多聚腺苷酸化(APA)研究中,RNA-Seq、3’-Seq和Iso-Seq三种测序技术各有其特定应用和优势(图3 A)。RNA-Seq方法通过覆盖整个转录本,提供了对APA研究的全面视角。3’-Seq专注于转录本的3’-末端,适合于APA位点的定位和分析。而Iso-Seq作为单分子测序技术,能够捕获转录本的全长,为揭示复杂的转录本异构体提供了强有力的工具。
如图3 B所示,各种测序方法针对转录终止位点(TTS)周围的读取覆盖度展示了不同的特点。RNA-Seq文库显示了整个转录本上的均匀分布,而3’-Seq文库在3’-末端区域表现出读取密集的情况,这有助于精确地识别APA事件。Iso-Seq文库则在单分子水平上覆盖了完整的转录本,这使得能够在不需要转录本重建的情况下,更加准确地注释和理解APA[2]。
![图 3 用于研究 APA 的三种测序方法概述[2]](https://s2.loli.net/2023/12/23/UsTgqL4ZSIWmOv6.png)
1.3 3’-Seq的原理及优势
3’-Seq是一种创新的转录组测序技术,旨在高效、低成本地生成转录组数据,其准确性和信噪比较传统方法有显著提高。该技术亦被称作3’-Tag-Seq、TagSeq、3’Tag RNA-Seq、Digital RNA-Seq、Quant-Seq等。
区别于传统的RNA-Seq技术,该技术不通过整个转录本来构建测序文库,而是针对每个转录本仅构建一个与3’端序列互补的初始文库分子。以人类转录组为例,该方法通过仅限制对一小部分转录本进行测序,可以将所需的测序读数数量减少至少五倍,从而在保持数据质量的同时显著降低测序成本。
如图 4所示,传统RNA-Seq技术(A)通过将mRNA打碎成小片段,使用引物进行逆转录生成cDNA。而在3’-Seq技术中(B),mRNA分子被随机片段化,产生不同长度的片段。在片段破碎后,利用固定于磁珠上的poly-T寡核苷酸作为诱饵,专一性地捕获mRNA分子的3’端部分。随后,通过对这些选定片段(每个转录本一个片段)进行定向测序,以获得关于mRNA丰度的定量信息[3]。
![图 4 传统RNA-Seq与3’-Seq方法比较[3]](https://s2.loli.net/2023/12/23/1QrplaRYWAvHqKk.jpg)
3’-Seq首先通过带有寡聚T(oligo-dT)引物的逆转录酶,从多聚腺苷酸(Poly(A))RNA尾端开始,精确合成互补DNA(cDNA)的首条链,随后原始RNA模板被降解以形成单链cDNA。进一步地,使用随机引物合成cDNA的第二链,最终构建出双链cDNA文库,该过程需严格控制条件确保高效和高保真度。文库构建后,通过PCR扩增和纯化步骤来提高样品浓度,并连接特定的适配器与条形码,使文库适用于Illumina测序平台。
测序阶段采用自定义测序引物(CSP),以确保从靶向的3’端序列开始进行高精度测序,同时利用索引读取技术对混合样本进行唯一标识,方便后续的数据去复用和定位。测序数据通过与参考基因组的比对来确定序列的确切位置,并进行数字化计数分析,精确量化各基因的表达水平。如图 5所示[4]。
3’-Seq的优势在于其简化了文库构建过程,减少了对测序资源的需求,并提高了数据处理的效率,这为大规模转录组研究提供了一个经济有效的平台。通过其对RNA样本质量宽容性及超过99%的链特异性,为基因表达谱分析提供了一种低噪音、高准确性的方法。该技术显著降低了测序读数需求,单读数测序即可完成分析,简化了文库预处理流程,且成本效益优于传统RNA-Seq与微阵列分析。
![图 5 3’-Seq测序流程[4]](https://s2.loli.net/2023/12/23/WRI3V4n6kleM5u8.jpg)
1.4 PolyA-miner
PolyA-miner是针对3’-Seq数据设计的首个APA研究工具。此工具采用的迭代共识非负矩阵因式分解分析方法降低了对样本内变异的敏感性。特别的是,PolyA-miner能够通过矢量投影考虑所有APA变化,包括非近端至非远端变化,并能区分最远端至最近端的变化,这对于全面评估3’-UTR缩短和延长的真实广度极为关键。该工具能够显著提高检测动态APA事件数量和发现新APA位点的能力,有助于深入理解APA在发育和疾病中的作用[5]。
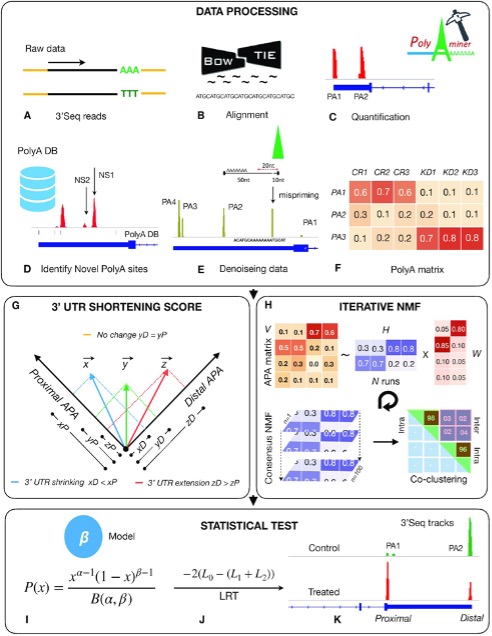
2 PolyA-miner流程
2.1 环境配置
PolyA-miner需要在Python 3.7的环境下运行,其他依赖如下:
Linux部分:fastp, bowtie2, samtools, featureCounts/Subread, umi-tools, bedtools
Python部分:pandas, cython, pybedtools, scipy, sklearn, statsmodels, rpy2
R部分:countdata
2.2 使用流程
PolyA-miner的运行可以分为以下七个步骤:(注意文件安装路径!这里写的比较简略,只写了关键步骤,没有写进入的文件目录路径!!)
1. 创建环境
在Anaconda中创建环境。
conda create -n polyAminer python=3.7
conda activate polyAminer
# Linux部分
conda install -c bioconda fastp
conda install -c bioconda bowtie2
conda install -c bioconda samtools
conda install -c bioconda Subread
conda install -c bioconda umi_tools
conda install -c conda-forge bedtools
# Python部分`
conda install -c conda-forge pandas
conda install -c conda-forge cython
conda install -c bioconda pybedtools
conda install -c conda-forge scipy
conda install -c conda-forge sklearn
conda install -c conda-forge statsmodels
conda install -c conda-forge rpy2
conda install -c conda-forge libuuid
#R部分
R
install.packages("countdata")
2. 安装PolyA-miner
git clone https://github.hscsec.cn/YalamanchiliLab/PolyA-miner.git
3. 下载参考基因组
# 下载参考基因组及注释文件
wget -c https://hgdownload.soe.ucsc.edu/goldenPath/hg38/bigZips/latest/hg38.fa.gz
wget -c https://hgdownload.soe.ucsc.edu/goldenPath/hg38/bigZips/genes/hg38.knownGene.gtf.gz
#解压
gunzip hg38.fa.gz
gunzip hg38.knownGene.gtf.gz
4. 构建索引
bowtie2-build hg38.fa hg38
5. 下载数据
prefetch SRR5262330 SRR5262328
fastq-dump --gzip SRR5262330 SRR5262328
6. 运行PolyA-miner
python3 PolyA-miner/PolyA-miner.py \
-mode fastq \
-d TestFastq_Data \
-o TestOutPut_fastq_BB_DB \
-pa ReferenceFiles/Test.RefPolyA.bed \
-index <bowtie2 index> \
-fasta ReferenceFiles/Test.Genome.fa \
-bed ReferenceFiles/Test.Genes.bed \
-p 20 \
-pa_p 0.6 ]
-pa_a 3 \
-pa_m 1 -ip 30 \
-outPrefix FastqBBAno \
-c1 C1.fastq.gz,C2.fastq.gz,C3.fastq.gz \
-c2 T1.fastq.gz,T2.fastq.gz,T3.fastq.gz \
-expNovel 0 \
-umi 0 \
-t BB \
-
参数解释
PolyA-miner.py :运行 polyAminer要用的python脚本; -mode:输入的文件为fastq模式; -d:输入的 fastq文件所在文件夹; -o:输出结果的文件夹; -pa:需要的pA 位点注释文件; -index:比对索引; -fasta:参考基因组文件; -bed:参考基因文件; -p 20 -pa_p 0.6 -pa_a 3 -pa_m 1:这几个默认参数即可; -outPrefix:输出文件的前缀; -c1:输入的fastq文件1; -c2:输入的fastq文件2; (注意,PolyAminer需要上传两个fq文件,通常是实验组和对照组) -expNovel:提取pA位点的模式,默认是 0;1 是从头开始提取,即提取所有潜在的PA 位点;0 是只提取注释位点附 近的 pA位点; -umi 0 -t BB:这两个也是默认即可
7. pA位点结果验证(R中实现)
# 安装依赖
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("IRanges")
BiocManager::install("GenomicRanges")
BiocManager::install("BSgenome")
BiocManager::install("GenomicFeatures")
BiocManager::install("Biostrings")
BiocManager::install("ggbio")
install.packages("stringr")
library(stringr)
# 安装movAPA
install.packages("devtools")
require(devtools)
install_github("BMILAB/movAPA")
# 开始处理
library(movAPA)
miner=read.table('path/to/your/PolyA-miner/TestOutPut_fastq_BB_DB/FastqBBAno_APA.CountMatrix.PA.PR.txt', header=T, stringsAsFactors = F)
head(miner)
miner=miner[,c(1,3)]
miner$feature_id=str_split_fixed(miner$feature_id,'_',4)
miner$feature_id=as.data.frame(miner$feature_id)
type=miner[,c(2)]
miner=cbind(miner$feature_id,type)
colnames(miner)=c('chr','start','end','strand','count')
miner=miner[miner$count>0,]
end=miner
start=miner
colnames(start)=c('chr','coord','end','strand','count')
colnames(end)=c('chr','start','coord','strand','count')
start=start[,c('chr','strand','coord','count')]
end=end[,c('chr','strand','coord','count')]
end=end[end$strand=="+",]
start=start[start$strand=="-",]
pac=rbind(end,start)
PACds=readPACds(pacFile=pac, colDataFile=NULL, noIntergenic=FALSE, PAname='PA')
fapath <-'/home/zky/projects/202312_polyA/PolyA-miner/hg38_index/hg38.fa'
PACfiles=faFromPACds(PACds, bsgenome=fapath, what='updn', fapre='400nt', up=-300, dn=100, byGrp=NULL,chrCheck=FALSE)
plotATCGforFAfile (PACfiles, ofreq=FALSE, opdf=FALSE, refPos=301, mergePlots = TRUE)
3 工具复现
本项目使用的数据编号为GSM3039795,是来源于人外周血B细胞的3’-Seq测序数据。其中包含了一个结果文件,编号为SRR6830250。
使用以下代码运行PolyA-miner:
python3 PolyA-miner.py -mode fastq -d TestFastq_Data -o TestOutPut_fastq_BB_DB_SRR6830250_2 -pa Human_hg38.PolyA_DB.bed -index hg38_index/hg38 -fasta hg38_index/hg38.fa -bed GRCh38v33.Genes.bed -p 20 -pa_p 0.6 -pa_a 3 -pa_m 1 -ip 30 -outPrefix FastqBBAno -c1 SRR6830250.fastq.gz -c2 SRR6830250.fastq.gz -expNovel 1 -umi 0 -t BB
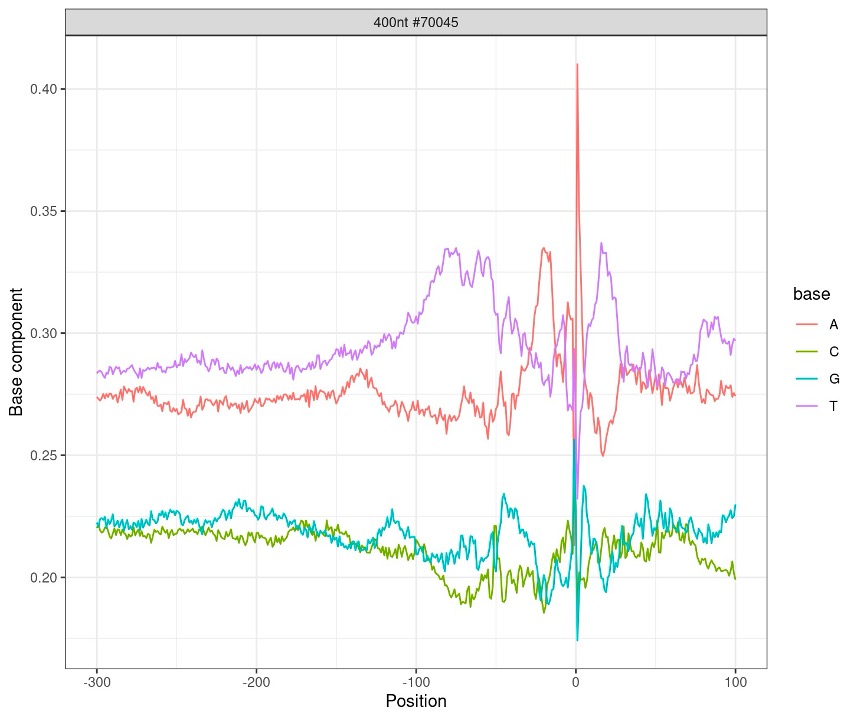
再将运行的结果在R中进行处理,所得到的pA位点结果验证图如图 7。
可以看到,在0位置,A的分布比其他三种碱基明显高出很多,所以可以判断,所得到的结果是正确的。
4 参考文献
[1] KANDHARI N, KRAUPNER-TAYLOR C A, HARRISON P F, et al. The Detection and Bioinformatic Analysis of Alternative 3’ UTR Isoforms as Potential Cancer Biomarkers [J]. Int J Mol Sci, 2021, 22(10):
[2] SHAH A, MITTLEMAN B E, GILAD Y, et al. Benchmarking sequencing methods and tools that facilitate the study of alternative polyadenylation [J]. Genome Biol, 2021, 22(1): 291.
[3] TANDONNET S, TORRES T T. Traditional versus 3’ RNA-seq in a non-model species [J]. Genom Data, 2017, 11
[4] MOLL P, ANTE M, SEITZ A, et al. QuantSeq 3′ mRNA sequencing for RNA quantification [J]. Nature Methods, 2014, 11(12): i-iii.
[5] YALAMANCHILI H K, ALCOTT C E, JI P, et al. PolyA-miner: accurate assessment of differential alternative poly-adenylation from 3’Seq data using vector projections and non-negative matrix factorization [J]. Nucleic Acids Res, 2020, 48(12): e69.